机器学习(ML)在材料化学中的应用的最新进展表明机器学习可以加速新材料的发现。在这篇文章中,Balaranjan Selvaratnam强调了ML在材料化学中的重要性。他们讨论了ML在合成、表征和预测材料活性方面的应用实例。最后,他们提出了这一领域的挑战,以及如何利用化学中ML进展与先进的机器人技术结合实现材料发现的自动化。
机器学习在材料合成方面的应用
化学领域研究人员发表的文献通常只包括反应成功的例子,但实际上大量未被报道的失败实验同样包含合成条件相关信息,这些失败实验包含的信息对预测反应成功和失败的边界条件也有重大价值。Racuglia等人使用ML方法,利用实验参数和试剂作为输入来确定模板化钒-亚硒酸盐晶体。他们利用实验室未成功的水热反应的数据训练机器学习模型,并用得到的模型来预测新的反应,所得的模型能够成功预测新的有机-无机材料的合成条件,合成成功率达89%。作者收集了大量实验室失败反应的数据,以反应物物理化学性能(如分子质量,元素周期表位置等)及反应条件(如反应物配比、反应温度、环境pH等)为特征,训练了一个SVM模型,该模型预测其测试集的反应结果时,准确率可达78%,对钒-亚硒酸盐体系反应的预测准确率达79%。通过将该SVM模型转换为方便人类理解的决策树模型,还能进一步认识反应相关机理,从而指导新的合成反应。

机器学习在材料表征方面的应用
利用衍射、电子显微镜、光谱学和其他技术对材料进行表征,可以了解材料的物理化学性质。利用粉末X射线衍射(PXRD)研究了多孔材料的相组成和有序性。Sohn和他的同事通过对模拟的XRD图谱进行训练,用卷积神经网络(CNN)来识别材料的相和组成。通过模拟含有Sr、Al、Li和O的结构的XRD获得数据。CNN模型经过训练之后可以识别材料的相和成分,然后,他们用模拟数据和真实数据的测试集对模型进行测试,对模拟测试数据的准确率大于99.5%,对真实数据的测试集的分类准确率大于97.3%。然后,利用CNN模型将回归问题转化为分类问题,预测每个相的百分比。这是通过预测成分的百分比对应的分类作为CNN模型的目标值来实现的,这实现了对未知混合物中的组成成分的近似估计。
机器学习用于预测催化反应
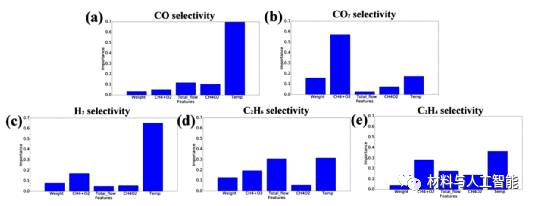
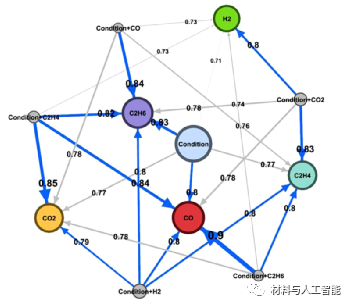
材料的应用要求材料朝着特定的性能和一定的活性阈值进行优化。因此,在实验之前预测材料的性质和活性是必要的。Takahashi和同事利用机器学习对196个实验甲烷氧化偶联反应(OCM)产物的选择性进行了研究,(1)建立了CO、CO2、C2H4、C2H6和H2选择性的预测模型,(2)构建OCM反应的反应网络。他们使用Mn-Na2WO4/SiO2作为催化剂,通过改变催化剂的质量、CH4 + O2的量、总流量、CH4/O2比和温度来收集数据。利用收集到的数据,他们构建了六个不同的机器学习模型:线性回归(LR)、支持向量机(SVM)、最小绝对收敛和选择算子(LASSO)、随机森林回归(RFR)和极限树回归(ETR)。通过超参数优化和交叉验证分析,RFR对预测任务表现出最佳性能。进一步分析了描述符对选择性预测的重要性。结果表明温度在CO和H2的选择性中起着重要的作用。CO2的选择性依赖于CH4 + O2的量,然而,C2H4和C2H6的选择性与几个实验参数有显著的相关性,如图2所示。为了理解OCM反应网络,他们训练了几个RFR模型,利用所有的实验参数和产品的选择性作为输入特征来预测每个产品的选择性。利用不同RFR模型的交叉验证得分建立反应路线图如图3所示。
以上例子表明了机器学习在驱动材料研究中的效用,ML可用于预测材料的各种性质和催化活性。然而,在使用ML模型时必须谨慎,因为它们可能过度拟合训练数据。为了让机器学习达到成熟阶段并加速发现新材料,他们提出可以将机器学习和与机器人技术结合实现新材料发现的自动化。
原文链接:https://doi.org/10.1016/j.cattod.2020.07.074





