上一篇:机器学习指南(一)
传统的机器学习
在接下来的内容,作者主要讨论了几种重要的机器学习方法,重点介绍它们的优缺点。表1显示了不同机器学习方法的比较。首先介绍的是,不基于神经网络的方法,也称为“传统机器学习”。此类模型可以使用各种软件包来训练,包括Python中的scikit-learn、R中的caret 和 Julia中的MLJ。
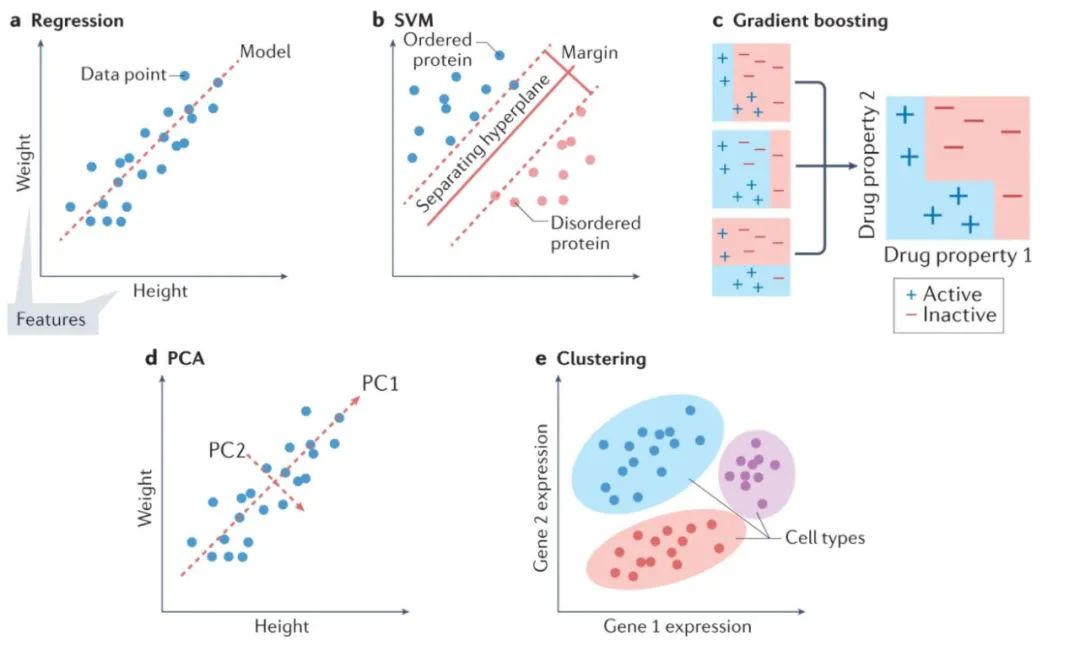
下图展示了传统机器学习的一些方法:
当开发用于生物学中,在给定任务寻找最合适方法时,通常会将传统机器学习视为第一个探索领域。深度学习就是其中一个例子。深度学习在其擅长的应用领域一般有三个要求:有大量可用数据(例如,百万个数据集)、每个数据点有很多个特征、特征高度结构化(特征彼此之间有明确的关系,例如图像中的相邻像素)。大部分的生物数据都可满足以上的要求,并且深度学习已成功应用到不同的生物数据,包括 DNA、RNA 和蛋白质序列,以及显微镜图像等数据。
然而,即使满足其它两个要求,对大量数据的要求也可能使深度学习不是最好的选择。与传统方法相比,深度学习开发和测试的速度比较慢。与支持向量机(SVM)和随机森林等传统模型相比,开发深度神经网络的架构然后对其进行训练可能是一项耗时且计算成本极高的任务。尽管存在一些方法,但对于深度神经网络,估计特征重要性(即每个特征对预测的重要性)或模型预测的置信度仍然不是一件容易的事,这两者在生物环境中通常是必不可少的。即使对于特定的,深度学习在技术上可行的生物预测任务,谨慎的做法通常还是:训练传统方法,再将其与基于神经网络的模型进行比较。
传统方法通常期望数据集中的每个例子都具有相同数量的特征,因此这并不总是可行的。一个明显的生物学示例是,在使用蛋白质、RNA 或 DNA 序列的时候,每个例子都具有不同的长度。要对这些数据使用传统方法,可以使用简单的技术(例如填充和加窗)更改数据,使它们的大小都相同。“填充”(padding)意味着对每个示例添加预测的值,直到它与数据集中最大的示例大小相同。相比之下,加窗(windowing)将单个示例缩短到给定的大小(例如,在一个序列长度至少为100的蛋白质序列数据集中,只使用每个蛋白质的前 100 个残基)。
使用分类和回归模型
对于如上图a所示的回归问题,回归(具有正则化项的线性回归)通常是构建模型的起点,因为它可以为给定任务提供快速且易于理解的基准。当期望模型依赖可用数据中的最少特征时,线性回归的其他形式,也值得考虑。但一般数据中特征之间的关系,通常是非线性的。对于这些情况,使用诸如SVM(support vector machine,支持向量机)之类的模型通常是更合适的选择。SVM 是一种强大的回归和分类模型,它使用核函数(kernel function)将不可分的问题转换为更容易解决的可分问题。SVM 可用于执行线性回归和非线性回归,具体取决于使用的核函数。开发模型的一个好方法是训练一个线性 SVM 和一个带有径向基函数核的 SVM(一种通用非线性类型的 SVM)来量化,以从非线性函数中获得的增益。非线性方法可以提供更强大的模型,但代价是不容易解释哪些特征影响模型。这就是前面所提到的偏差-方差权衡。
回归中常用的许多模型也可用于分类。训练线性 SVM 和带有径向基函数核的 SVM 也是分类任务的一个很好的默认起点。另一种可以尝试的方法是k 最近邻(k nearest neighbours)分类。作为最简单的分类方法之一,k 最近邻分类提供了一个有用的基线性能标记,可以与其他更复杂的模型(例如 SVM)进行比较。另一类稳健的非线性方法是基于集成(ensemble)的模型,例如随机森林和 XGBoost。这两种方法都是非常强大的非线性模型,好处在于能额外提供特征重要性的估计,并且通常需要最少的超参数调整。如果某个生物学任务需要了解特征对预测的贡献,那么这些模型就是一个不错的选择,毕竟它们能分配特征重要性值,决策树结构。
对于分类和回归,许多可用的模型往往具有令人眼花缭乱的特点。试图先验地预测特定方法对特定问题的适用程度可能不适合,而采用经验、反复试验的方法来寻找最佳模型通常是最谨慎的方法。使用现代机器学习函数——例如 scikit-learn——在这些模型之间进行更改,通常只需要更改一行代码。因此,选择最佳方法的策略是:训练和优化上述各种方法,选择在验证集上性能最好的那个,最终比较它们在单独的测试集上的性能。
使用聚类模型。聚类算法的使用在生物学中很普遍。k平均(k-means)是一种强大的通用聚类方法,与许多其他聚类算法一样,它需要将聚类数设置为超参数。DBSCAN 是一种替代方法,它不需要预定义集群的数量,但是需要设置其他超参数。也可以在聚类之前执行降维,以提高在具有大量特征的数据集上的性能。
降维
降维(dimensionality reduction)用于将具有大量属性(或维度)的数据转换为低维形式,同时尽可能保留数据点之间的不同关系。例如,相似的数据点(例如,两个同源蛋白质序列)在其低维形式上也应该相似,而不同的数据点(例如,不相关的蛋白质序列)应该保持不相似。。通常选择两个或三个维度,来可视化数据,尽管更多的维度也用于机器学习。这些技术包括数据的线性和非线性变换。生物学中常用的例子包括主成分分析(principle component analysis,PCA, 上图d)、UMAP(Uniform Manifold Approximation and Projection)和t-SNE(t-distributed stochastic neighbour embedding)。生物学数据处理中具体使用的技术取决于情况:PCA 保留数据点之间的全局关系并且是可解释的,因为每个组件都是一个输入特征的线性组合,这意味着很容易理解哪些特征会导致数据的多样性。t-SNE 更好地保留了数据点之间的局部关系,是一种灵活的方法,可以揭示复杂数据集中的结构。应用包括用于 t-SNE的单细胞转录组学和用于主成分分析的分子动力学轨迹分析。
今天的介绍到这里就结束了,希望大家有所收获,下期会继续给大家更新人工神经网络的知识。
参考文章:
A guide to machine learning for biologists
https://doi.org/10.1038/s41580-021-00407-0





